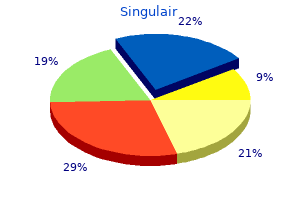
Singulair 4 mg amex
Several different mutations have been reported in families during which extreme thrombocytosis has been attributed to increased thrombopoietin manufacturing asthma treatment nz singulair 10 mg order without prescription. The thrombopoietin receptor in the Dutch family was normal asthma 7 month old baby 10 mg singulair order free shipping, yet there was a G-to-C transversion in the splice donor website of intron three of the thrombopoietin gene. All of the affected members of the Dutch family were shown to have elevated thrombopoietin ranges. In this family, some extent mutation in the thrombopoietin gene was believed to lead to systemic overproduction of thrombopoietin, leading to a familial form of thrombocytosis. Additional households with thrombocytosis caused by an analogous genetic mechanism have been recognized. The polymorphism happens completely in African�Americans and seems to have an autosomal dominant sample of inheritance with incomplete penetrance as a outcome of some heterozygotes have normal platelet counts whereas others have thrombocytosis. It leads to extreme thrombocytosis in homozygotes and sometimes to mild thrombocytosis in heterozygotes. In the households described with this germ-line mutation, excessive thrombocytosis was related to homozygosity and the mode of inheritance is considered being autosomal recessive with possible gentle manifestations occurring in heterozygotes. These problems have been initially thought-about to be associated with a benign scientific course, however follow-up of such families for longer durations of time has corrected this misperception. Also, these members of the family experienced vasomotor signs, including erythromelalgia and Raynaud phenomena, which responded to aspirin therapy however not hydroxyurea therapy. The affected person with acute leukemia had not obtained any chemotherapeutic agents, and analysis of the strength of the connection between this high thrombopoietin condition and the event of acute leukemia requires further investigation of those families. Occasional sufferers with thrombocytosis and elevated ringed sideroblasts but with out anemia have also been described. Unfortunately, techniques to examine clonality are presently not widely available and are restricted to the analysis of female patients. Such studies may be particularly helpful in young feminine patients with thrombocytosis. Probes for a variety of genes on the X chromosome can be informative for clonal evaluation of blood cell manufacturing in additional than 72% of feminine Americans. Polyclonal hematopoiesis is found in all circumstances of reactive and familial thrombocytosis. Initial studies, nevertheless, have advised that girls with polyclonal hematopoiesis could have fewer thrombotic complications than those with clonal hematopoiesis. In an asymptomatic affected person, follow-up to decide whether or not the diploma of thrombocytosis increases is warranted. If extra clues to the cause of the thrombocytosis are subsequently revealed, a diagnosis will become obvious. Some reassurance is provided by reviews of larger cohorts of patients, every with platelet counts of greater than a thousand � 109/L, which have been followed for years. Virtually not certainly one of the sufferers with reactive thrombocytosis developed a cerebrovascular accident, thrombophlebitis, or a peripheral arterial thrombosis. However, a research of 322 consecutive sufferers seen on the Mayo Clinic and adopted for a median follow-up of thirteen. Multivariable analysis identified an age at analysis of 60 years or older, leukocytosis (>15,000/�l), earlier venous thrombosis, tobacco use, and diabetes mellitus as unbiased predictors of poor survival. The rate of leukemic transformation was larger in patients with platelet counts above 1000 � 109/L and abnormal hemoglobin levels. Analysis of this Chapter69 EssentialThrombocythemia Overall survival (proportion) 1. Low-risk (0�1 points), intermediate-risk (2 points), and high-risk (3+) categories have been associated with 1. The phenotype of the blast cell could be myeloid, myelomonocytic, megakaryocytic, of blended lineage, or even lymphoblastic. The danger of growing acute leukemia after remedy with hydroxyurea alone has been reported to enhance only barely (3�4%), but the sequential use of hydroxyurea with different cytotoxic brokers, similar to busulfan or pipobroman, significantly will increase the chance of creating a secondary leukemia. The development of acute leukemia is often related to a deletion of the quick arm of chromosome 17, which is most frequently deleted in hydroxyurea-treated patients, but a trisomy of the long arm of chromosome 1 and monosomy 7q has been observed in patients treated with pipobroman. These cytogenetic abnormalities are believed to be induced by means of these chemotherapeutic agents. The median survival after the event of myelodysplasia or leukemic transformation is 4 months. Because allogeneic stem cell transplantation could be healing, speedy referral to a transplant heart is recommended if the patient has an acceptable performance standing and an allogeneic stem cell donor is available. Both age (>60 years of age) and history of a previous thrombosis are widely accepted predictors of a patient developing extra thrombotic events throughout follow-up (Table 69. Other predictors of cardiovascular morbidity embody a historical past of smoking, hypertension, weight problems, diabetes mellitus, and congestive coronary heart failure, and a white blood cell depend larger than 11 � 109/L. Ironically, patients with platelet counts larger than 1000 � 109/L have a lower threat of creating an arterial thrombosis and a better risk of bleeding. Such stratification methods have been used to make treatment decisions to reduce the platelet rely. Using such patient-stratification methods, patients have been positioned into high-, intermediate-, or low-risk teams based on their predicted threat of creating an additional life-threatening thrombotic event. This study concerned a total of 114 patients, and the median follow-up interval was only 27 months. In reality, in several studies, an elevated leukocyte rely of above 11 � 109/L has been extra carefully related to the danger of creating additional thromboses than the diploma of elevation of the platelet rely. These conflicting reports in the literature among consultants in this area make it increasingly harder to be dogmatic about who to treat with platelet-lowering agents and what the target platelet depend ought to be. If a patient of any age has extreme thrombocytosis and a thrombotic or hemorrhagic occasion, that affected person have to be handled. Life-threatening thrombotic occasions require platelet pheresis together with the institution of myelosuppressive therapy. In this case, immediate physical removing of large numbers of platelets is most well-liked as a result of chemotherapeutic brokers generally require 18�20 days earlier than platelet counts could be decreased to normal levels. It is really helpful to cut back the platelet rely to 500,000/mm3 by each platelet pheresis and advised that achievement of such a goal requires the passage of two blood volumes over a 3�4-hour period. Such a therapeutic approach has been used to deal with acutely ill sufferers with problems such as cerebrovascular accidents, myocardial infarction, transient ischemic assaults, or life-threatening gastrointestinal hemorrhage. Long-term platelet pheresis is an ineffective means of controlling thrombocytosis, presumably because of the fast price of production of platelets. Therefore, most clinicians begin by administering a chemotherapeutic agent that has a rapid onset of action, similar to hydroxyurea at doses of 2�4 g/day, concurrently with the establishment of platelet pheresis. The dose of hydroxyurea requires close monitoring with acceptable reduction of dose to avoid excessive myelosuppression. Most investigators try to normalize the platelet count or reach a platelet rely at which the signs of the high-risk patient resolve. Although major bleeding episodes requiring hospitalizations are uncommon, patients with extreme thrombocytosis (>1500 � 109/L), acquired von Willebrand syndrome, and historical past of hemorrhagic episode are clearly at risk for developing further bleeding complications.
Discount 5 mg singulair mastercard
Hydroxyurea is used in the remedy of myeloproliferative neoplasms together with important thrombocythemia asthma symptoms preschoolers singulair 5 mg with mastercard, polycythemia vera and myelofibrosis (Chapters 68�70) asthma treatment xolair singulair 5 mg amex. These brokers bind thymidylate synthase and dihydrofolate reductase, and disrupt cell cycle progression and cell division. Methotrexate is the agent currently used in hematologic illnesses, whereas the household of brokers contains pemetrexed and trimetrexate. Inhibition of folatedependent methyl transfer enzymes disrupts purine synthesis pathways. Since it has no tumor selectivity, normal tissues in energetic cell cycle are affected, including mucosa, bone marrow and hair follicles, leading to mucositis, pancytopenia and alopecia. A third mechanism is decreased thymidylate synthase, and the fourth mechanism of resistance is impaired methotrexate polyglutamate formation. In quickly replicating cells, it stalls polymerase operate, leading to replication fork collapse and strand breaks on the replication fork, signaling apoptosis and differentiation. Specific resistance could be because of nucleotide transport down regulation, uncommon in dividing cells. Low penetration into the cerebrospinal fluid is overcome by bolus administration of very high doses: 1�2 g/m2 over 1 hour. Many modifications to these schedules have improved tolerance without sacrificing efficacy. Specific resistance is brought on by upregulation of deoxycytidine deaminase, which metabolizes gemcitabine to 2,2-difluorodeoxyuridine. Nonspecific resistance emerges because of upregulation of membrane transporters, though their function in clinical resistance is less clear. The combination of oxaliplatinum or cisplatin and gemcitabine is effective and nicely tolerated, and nearly all of sufferers remain eligible for autologous stem cell assortment and transplantation. Alternatively, they block key enzymes in de novo purine or pyrimidine biosynthesis. These brokers are predominantly cycle-active brokers and generally are phase specific, being primarily active in opposition to cells in S section. Because the expansion fraction of hematologic malignancies tends to be greater than that of nonhematologic malignancies, nucleoside analogs are notably helpful within the former problems. In contrast to alkylating agents, nucleoside analogs have restricted carcinogenic and leukemogenic potential. It is recognized for its immunosuppressive operate and inducing tolerance to allografts transplantation. Fludarabine phosphate is the 2-fluoro, 5-monophosphate spinoff of vidarabine (9-D-arabinofuranosyladenine [ara-A]) and is converted to the di- and triphosphate by intracellular kinases, as are gemcitabine and ara-C. Like different nucleoside analogues, it causes replication fork collapse, doublestrand breaks, and induction of P53, leading to apoptotic signaling. Its efficacy in opposition to normal lymphoid T and B cells seems linked to each cytotoxicity towards proliferating cells and resting cells; the latter effect is mediated by interference with the normal exercise of the nucleotide excision restore pathway. Fludarabine has also been utilized in refractory leukemias, marginal cell and other low-grade lymphomas. Recognition of the looks of lymphopenia after remedy led to research establishing fludarabine as part of the nonmyelosuppressive preparative regimens for allogeneic transplantation, used particularly for older individual recipients. This is most frequently at a dose of 30�35 mg/m2 for 5 days and used in combination with radiation, melphalan, or cyclophosphamide. In addition to lymphopenia, a quantity of cycles are related to extended myelosuppression and a continual peripheral neuropathy. Reversible liver toxicity and myelosuppression could be dose limiting (see Chapter 59). Before describing the precise inhibitors, a quick evaluation of the drug targets (topoisomerase enzymes) will be introduced (Table fifty seven. Topoisomerase I is catalytically active as a 100-kDa Clofarabine (2-chloro-2-arabino-flouro-2-deoxyadenosine) is a purine analog with exercise in sufferers with relapsed acute leukemia. Its activation requires cellular uptake and conversion to the triphosphate nucleotide. Early scientific studies with camptothecin had been stopped primarily due to hemorrhagic cystitis resulting from conversion of the sodium salt type to the active lactone form owing to its acidic pH within the bladder. Renewed curiosity in camptothecin occurred in 1985 when topoisomerase I was recognized as the target of this drug and as new extra water-soluble analogs grew to become obtainable. The fourth resistance mechanism involves alterations in the subcellular distribution of the enzyme. PlatinumAnalogs Mechanism of Action During a examine of the consequences of electrical current on growing bacteria, the antibacterial and, later, the antitumor actions of the platinum compounds have been fortuitously discovered. The antitumor agent cisplatin, its cis-carboxylester analog, carboplatin, and the diaminocyclohexane-containing oxaliplatin, are heavy-metal platinum complexes. MiscellaneousAgents Among the brokers included on this class, solely plicamycin, bleomycin, procarbazine, L-asparaginase, gallium nitrate, and glucocorticoids are of present curiosity to hematologists; these are mentioned in Appendix 57. Dobbelstein and Moll describe three "waves" or "epochs" in anticancer drug development. Second-wave drugs target mobile indicators, including those mediated by surface receptors and protein kinases. The targets of signaling inhibitors are diverse, and embody the products of oncogenes, essential for the event and survival of neoplasms ("oncogene addition"). This state is named "nononcogene dependancy" and has expanded the variety of targets that can be used for most cancers treatment with signaling inhibitors. The third wave of anticancer drugs target cellular mechanisms and effector methods distinct from those focused by medication of the first two waves, but that are nonetheless essential for the survival of cancer cells. Other agents not easily included in these three waves embrace immunomodulatory drugs (thalidomide, lenalidomide, and pomalidomide), as properly as agents promoting cancer cell differentiation (all-trans retinoic acid). Many of those mutations are present at the time of prognosis and resistant clones emerge after exposure to imatinib. Common antagonistic events because of imatinib include fluid overload and edema, and development of coronary heart failure, fatigue, rash, and myelosuppression. Gastrointestinal side effects are widespread, and nausea and vomiting are frequent causes for poor compliance. This lowered structural stringency in kinase inhibition allows for inhibition of all kinase mutations, excluding the T315I mutation. As a multikinase inhibitor, dasatinib has a broader and alternative toxicity profile, with pulmonary edema, pleural effusions, and thrombocytopenia being the more frequent antagonistic results. Recently, greater charges of pulmonary hypertension have been noticed in patients receiving dasatinib. Common adverse events with nilotinib include rash, gastrointestinal disturbances (nausea, vomiting, diarrhea), in addition to neutropenia and thrombocytopenia.
Best 4 mg singulair
Summary of pink cell enzyme defects in descending order of their medical significance asthma treatment guidelines stepwise order 5 mg singulair with mastercard. This evaluate focuses on the impression of vitality metabolism of erythrocyte and its pathophysiology asthma severity singulair 4 mg with mastercard. Viprakasit V, Ekwattanakit S, Riolueang S, et al: Mutations in Kruppellike factor 1 trigger transfusion-dependent hemolytic anemia and persistence of embryonic globin gene expression. Review of red cell enzymes with intensive bibliography of original and up to date articles. In sufferers with severely dysfunctional spectrin mutations, the weakened spectrin dimer-dimer self-association disrupts the skeletal lattice, resulting in a marked skeletal instability and cell fragments. It is speculated that elliptocytes are completely deformed cells as a outcome of the weakened horizontal interactions facilitate a shear stress-induced rearrangement of skeletal proteins, precluding recovery of the normal biconcave shape. Acanthocytosis,Stomatocytosis,andtheBilayer CoupleHypothesis the mechanism of acanthocytosis and stomatocytosis related to defects of membrane proteins is way much less clear. Most types of acanthocytosis are related to both acquired or inherited abnormalities of membrane lipids. In rare cases with acanthocytosis, membrane protein abnormalities have been detected, but the associated mechanisms leading to acanthocyte formation are unknown. These abnormalities occur within the McLeod phenotype, the choreaacanthocytosis syndrome, and other uncommon issues. In acanthocytosis erythrocytes, agents that work together with the lipids of the inner lipid bilayer leaflet normalize the form. Vertical interactions, that are perpendicular to the airplane of the membrane, stabilize the lipid bilayer. These interactions embody spectrinankyrin�band three interactions, spectrin-protein 4. Horizontal interactions, that are parallel to the aircraft of the membrane, help the structural integrity of erythrocytes after their publicity to shear stress. Horizontal interactions involve the spectrin heterodimer association web site, the place spectrin heterodimers assemble into tetramers, the principal building blocks of the membrane skeleton, and the contacts of the distal ends of spectrin heterodimers with actin and protein four. Although interactions between proteins of the erythrocyte membrane are significantly extra complex than can be categorised by horizontal and vertical interactions, the model serves as a helpful beginning place for understanding erythrocyte membrane protein interactions, notably in reference to membrane-related issues. Consequently, the lipid bilayer membrane is destabilized, leading to release of bilayer lipids from the cells in the type of skeleton-free lipid vesicles. This lipid loss, in flip, results in membrane surface space deficiency and spherocytosis. The lipid bilayer forms the equator of the cross-section with its polar heads (small circles) turned outward. Mutations in the extremely conserved region of -spectrin involved within the interplay with protein 4. Thus sufferers with one normal and one faulty -spectrin allele are asymptomatic, because -spectrin manufacturing remains in extra of -spectrin synthesis, allowing normal amounts of spectrin heterodimers to be assembled on the membrane. Destabilization of the lipid bilayer facilitates a release of lipids from the membrane, leading to surface area deficiency and formation of poorly deformable spherocytes that are selectively retained and damaged in the spleen. This facilitates disruption of existing protein contacts during shear stress-induced elliptical deformation. In some circumstances, mutations of the ankyrin promoter resulting in decreased ankyrin expression have been discovered. Approximately 15% to 20% of ankyrin gene mutations reported are de novo mutations. Ankyrin deletions could also be part of a contiguous gene syndrome with manifestations of spherocytosis, mental retardation, typical facies, and hypogonadism. A variety of band 3 mutations clustered within the membranespanning domain that replace extremely conserved arginines have been described. These arginines, which are all positioned on the cytoplasmic end of a predicted transmembrane helix, exhibit defective mobile trafficking from the endoplasmic reticulum to the plasma membrane. Chapter45 RedBloodCellMembraneDisorders 629 Spectrin/ankyrin deficiency Release of microvesicles pH Macrophage contact Hemolysis Band 3/protein four. The main defect in hereditary spherocytosis is a deficiency of membrane surface area. Decreased surface space could additionally be produced by two completely different mechanisms: (1) Defects of spectrin, ankyrin, or protein four. Both pathways end in membrane loss, decreased surface space, and formation of spherocytes with decreased deformability. These deformed erythrocytes turn out to be trapped within the hostile surroundings of the spleen the place splenic conditioning inflicts additional membrane harm, amplifying the cycle of purple cell membrane injury. Alleles have been recognized that influence band 3 expression and that, when inherited in trans to a band three mutation, aggravate band three deficiency and worsen the scientific severity of the illness. The lack of membrane material happens via the discharge of vesicles containing integral proteins devoid of spectrin. During in vitro incubation, the loss of membrane materials is sufficient to increase the floor space deficiency, as evidenced by increased osmotic fragility of the cells after incubation. Another attainable mechanism could contain a formation of band 3-free domains in the membrane, followed by the formation of membrane blebs, which are subsequently released from the cells as microvesicles. Such a speculation relies on the remark that aggregation of intramembrane particles (composed principally of band 3) in ghosts results in the formation of particle-depleted domains from which membrane lipids bleb off as microvesicles. Additional evidence supporting the latter mannequin comes from the band three knock-out mouse model and from human, cow, and zebrafish instances of complete band 3 deficiency. Erythrocytes lacking band three spontaneously shed membrane vesicles, leading to spherocytosis and hemolysis. The mobile dehydration may be attributable to activation of pathways causing a selective lack of potassium and water or a hyperactive Na+/K+ pump. Normal discocytes have an excess floor, which allows them to deform and move through slender microcirculation openings. Consequently, the nondeformable spherocytes accumulate within the purple pulp, which turns into grossly engorged. Anemia is usually mild to moderate but could additionally be absent due to compensatory bone marrow hyperplasia manifest by reticulocytosis. Splenomegaly gradually develops in most patients, with the spleen sometimes reaching giant dimensions. These mice develop a neurologic syndrome with a development that coincides with the lack of ankyrin from the Purkinje cells of the cerebellum. The remaining circumstances characterized by a recessive inheritance pattern are caused by a defect in protein 4. These sufferers have a severe hemolytic anemia, whereas their largely consanguineous parents have a gentle to reasonable form of the disease or are asymptomatic. These hyperdense erythrocytes can be detected with newer laser-based blood counters or using aperture impedance evaluation obtainable in lots of scientific laboratories. However, these abnormalities can be absent in people with a gentle type of the disease. Frequent spheroovalocytes and stomatocytes have been reported in Japanese patients with protein four. Thus the cell behaves as an almost perfect osmometer in that it will increase its volume in hypotonic solutions progressively till a "critical hemolytic quantity" is reached.
4 mg singulair discount free shipping
DeSimone J definition of asthma gina order 4 mg singulair with amex, Koshy M asthma definition ubiquitous buy 4 mg singulair with visa, Dorn L, et al: Maintenance of elevated fetal hemoglobin levels by decitabine during dose interval treatment of sickle cell anemia. May C, Rivella S, Callegari J, et al: Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human betaglobin. Rivella S, May C, Chadburn A, et al: A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. May C, Rivella S, Chadburn A, et al: Successful treatment of murine beta-thalassemia intermedia by transfer of the human beta-globin gene. Lacerra G, Sierakowska H, Carestia C, et al: Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients. Baum C, Dullmann J, Li Z, et al: Side results of retroviral gene transfer into hematopoietic stem cells. Breda L, Gambari R, Rivella S: Gene therapy in thalassemia and hemoglobinopathies. Takahashi K, Yamanaka S: Induction of pluripotent stem cells from mouse embryonic and grownup fibroblast cultures by outlined components. Okita K, Ichisaka T, Yamanaka S: Generation of germline-competent induced pluripotent stem cells. Aksoy M, Bermek E, Almis G, et al: beta-Thalassemia intermedia homozygous for normal hemoglobin A2 beta-thalassemia. Sbyrakis S, Karagiorga-Lagana M, Voskaki I, et al: A easy index for initiating transfusion therapy in thalassaemia intermedia. Aessopos A, Kati M, Meletis J: Thalassemia intermedia today: ought to patients often receive transfusions Cossu P, Toccafondi C, Vardeu F, et al: Iron overload and desferrioxamine chelation therapy in beta-thalassemia intermedia. Borgna Pignatti C, Carnelli V, Caruso V, et al: Thromboembolic events in beta thalassemia major: an Italian multicenter research. Aessopos A, Farmakis D, Karagiorga M, et al: Cardiac involvement in thalassemia intermedia: a multicenter examine. Saisorn I, Leewansangtong S, Sukpanichnant S, et al: Intrarenal extramedullary hematopoiesis as a renal mass in a patient with thalassemia. Aarabi B, Haghshenas M, Rakeii V: Visual failure brought on by suprasellar extramedullary hematopoiesis in beta thalassemia: case report. Ibabao J, Kassapidis S, Demetis S, et al: Bilateral pleural effusions in a beta-thalassemia intermedia patient with posterior mediastinal extramedullary hematopoietic masses. Kapelushnik J, Shalev H, Schulman H, et al: Upper airway obstructionrelated sleep apnea in a toddler with thalassemia intermedia. Taher A, Skouri H, Jaber W, et al: Extramedullary hematopoiesis in a patient with beta-thalassemia intermedia manifesting as symptomatic pleural effusion. Sorcinelli R, Cacace E, Del Piano M: Optic nerve compression by extramedullary hematopoietic tissue in a patient suffering from betathalassemia intermedia. Silvestroni E, Bianco I: A highly price efficient method of mass screening for thalassaemia. Carr S, Rubin L, Dixon D, et al: Intrauterine therapy for homozygous alpha-thalassemia. Naqvi J, Morrow W, Nisbet-Brown E, et al: Normal development of an infant with homozygous alpha thalassemia [abstract]. Fucharoen S, Winichagoon P: Clinical and hematologic aspects of hemoglobin E beta-thalassemia. An unstable beta-chain variant producing the phenotype of extreme beta-thalassemia. Vercellotti Since it was acknowledged as the "first molecular disease," sickle cell anemia brought on by homozygosity for the mutant sickle beta globin gene has offered the basic paradigm for single-gene issues. Predominant scientific features include hemolytic anemia, episodic painful occasions, continual organ deterioration, disparate acute and continual issues, and a foreshortened life span. This article addresses the pathophysiology that underlies the sickle cell disease syndromes described in Chapter forty two. Each designation refers to an ethnographic region during which the sickle mutation achieved excessive gene frequency (typically peaking at zero. Origin,Selection,andDispersionoftheSickleGene the residence of each A and S alleles on the distinct regional cluster haplotypes means that the sickle mutation arose independently in the 5 areas. Historical and biologic data argue that frequency of the S gene tremendously expanded in Africa about 3000 years ago and in South Asia about 4000 years ago, following the introduction of iron tools. That led to adoption of an agricultural system that promoted both elevated human habitation density and favorable breeding situations for the mosquito vector, Anopheles, which in flip enabled growth of endemic Plasmodium falciparum. However, these with sickle trait are much less prone to develop high-level parasitemia or to have extreme malaria, an effect largely exerted early in childhood. One proposed mechanism hyperlinks protection to the instability of HbS, immune status, and splenic operate. Yet, the blunted malarial susceptibility in sickle trait displays a fancy interrelationship among the sickle gene, host biology, and environmental components. Eventually, the sickle gene spread geographically by means of commerce, migration, and the slave trade. In 1949, Neel validated the Mendelian autosomal dominant inheritance of sickle cell anemia, and Pauling demonstrated presence of an abnormal hemoglobin (Hb) in sufferers and carriers. This was followed by remark of the poor solubility of deoxygenated sickle Hb (HbS) and the reversible sol-gel transformation of HbS options. Thereafter, more and more detailed investigations began to reveal the putting complexities of sickle cell disease pathobiology. Genes for different -globin variants are allelic to the S gene and have a codominant impact. Worldwide, about 75% of sickle cell anemia births now occur in sub-Saharan Africa, 15% in India, 5% within the Americans, 4% in the Eastern Mediterranean, 1% in Europe. Blood smears prepared beneath differing situations, using antecubital blood from the identical sickle cell anemia affected person. Most cells resumed normal form, but one elongated, irreversibly sickled cell stays present. The five regions during which the sickle gene achieved excessive allelic frequency are superimposed on shading that identifies the Old World distribution of the sickle gene and of historic, endemic malaria. As a generalization, it unfold on the Benin haplotype to North Africa and then throughout the Mediterranean. All three major African haplotypes are present within the western Arabian Peninsula; however on the eastern side, the sickle gene tends to be on the Arab-India haplotype. This can additionally be true in India, though sub-Saharan haplotypes are represented as well.
Singulair 5 mg buy free shipping
While pores and skin is the commonest presenting website asthmatic bronchitis japanese discount singulair 5 mg amex, lymphomas of gamma delta T-cell origin can present in other primarily extranodal sites asthma treatment plan singulair 5 mg generic mastercard. Although sufferers might respond initially to chemotherapy, relapse has been seen within the overwhelming majority of instances, and the median survival is less than three years. Rare long-term survival has been seen following allogeneic hematopoietic cell transplantation. The pattern of infiltration mimics the homing sample of gamma-delta T cells with marked sinusoidal infiltration in liver and spleen. Abnormal cells are often present within the sinusoids of the bone marrow however may be tough to identify without immunohistochemical stains. The neoplastic cells also have a phenotype that resembles that of regular resting gamma-delta T cells. Isochromosome 7q is a consistent cytogenetic abnormality, and is often seen in affiliation with trisomy eight. All are seen most frequently in Asian kids however are also reported in Central and South America, in individuals of Native American origin. The latter two situations affect primarily the skin and have a more indolent medical course, whereas the systemic disease has a very aggressive scientific course with survival measured in weeks. HodgkinLymphomas Hodgkin and non-Hodgkin lymphoma have long been considered distinct illness entities primarily based on their differences in pathology, phenotype, scientific options, and response to remedy. In addition, the presence of somatic mutations indicates transit by way of the germinal center. The most popular time period of Hodgkin lymphoma over Hodgkin illness reflects present information concerning the nature of the neoplastic cell as a lymphocyte. It impacts adults (median age 50) and the commonest scientific presentation is a harmful nasal or midline facial lesion. The medical course is normally aggressive, with a barely improved median survival in patients with localized disease, in which local radiation therapy may be helpful. Low-power illustration reveals vague expansile nodules that efface the lymph node structure (A). Progressively reworked germinal facilities are sometimes seen in partially concerned lymph nodes or other lymph node websites. The background is predominantly lymphocytes with or without epithelioid histiocyte clusters. Small lymphocytes in the nodules are predominantly B cells with a mantlezone phenotype. Patients with superior stage illness might profit from remedy regimens used for aggressive B-cell lymphomas. Progression to a course of resembling T-cell/histiocyte wealthy massive B-cell lymphoma may been seen, and recent information recommend that these illnesses could also be different ends of a spectrum, with a detailed biologic relationship. The background contains lymphocytes, histiocytes, plasma cells, eosinophils, and neutrophils. Classic Hodgkin Lymphoma, Mixed Cellularity Patients are often adults; males outnumber females and the stage is often superior. It incessantly presents with belly lymphadenopathy, spleen, liver, and bone marrow involvement, with out peripheral adenopathy. The infiltrate is diffuse and sometimes seems hypocellular, owing to the presence of diffuse fibrosis and necrosis. Classic Hodgkin Lymphoma, Nodular Sclerosis this variant is most common in adolescents and younger adults, but can occur at any age; female circumstances equal or exceed those in males. In nodular sclerosis Hodgkin Lymphoma, broad bands of sclerosis usually divide the lymph node into cellular nodules (A). The nodules comprise a combined mobile infiltrate and scattered neoplastic cells with lobular nuclei and retracted cytoplasm (B). In blended cellularity Hodgkin lymphoma, the lymph node is normally diffusely effaced with only nice fibrosis (C). Classic mononuclear, binuclear, and multinuclear Hodgkin and Reed-Sternberg cells are present (D). Hallek M: Chronic lymphocytic leukemia: 2015 update on analysis, risk stratification, and therapy. Horn H, Schmelter C, Leich E, et al: Follicular lymphoma grade 3B is a distinct neoplasm based on cytogenetic and immunohistochemical profiles. Van Loo P, Tousseyn T, Vanhentenrijk V, et al: T-cell/histiocyte-rich giant B-cell lymphoma reveals transcriptional features suggestive of a tolerogenic host immune response. Vater I, Montesinos-Rongen M, Schlesner M, et al: the mutational sample of major lymphoma of the central nervous system decided by whole-exome sequencing. Vose J, Armitage J, Weisenburger D: International peripheral T-cell and pure killer/T-cell lymphoma research: pathology findings and medical outcomes. Hartmann S, Doring C, Jakobus C, et al: Nodular lymphocyte predominant Hodgkin lymphoma and T cell/histiocyte wealthy massive B cell lymphoma - endpoints of a spectrum of one disease Thus the molecular evaluation of these cells was hampered until methods grew to become obtainable to isolate these cells by microdissection from tissue sections. Hence it was initially tough to draw agency conclusions from the study of such lines. B cells are generated in the bone marrow from hematopoietic stem cells in a multistep developmental course of. B-cell growth is initiated when widespread lymphoid progenitors bear gene rearrangements on the Ig gene heavy chain locus. The variable part of the antibody heavy chain is composed of three gene segments: variable (V), range (D), and becoming a member of (J). A heavy chain can be expressed and the developmental stage of a pre-B cell is reached if the rearrangement is in-frame and productive. Moreover, because of the provision of a quantity of V, D, and J gene segments and additional variety generated on the becoming a member of sites of the rearranging gene segments, a V(D)J rearrangement (in specific for the heavy chain locus) is exclusive for every B cell and thus can be used as a clonal marker for B cells deriving from the same mature B cell. The process of somatic hypermutation introduces point mutations and a few deletions and duplications at a really high price into the Ig heavy and lightweight chain V region genes. In class switching, the originally expressed C� and C heavy chain fixed area genes (encoding IgM and IgD, respectively) are replaced by downstream positioned C, C, or C genes, encoding IgG, IgA, and IgE heavy chains, respectively, so that antibodies with altered effector capabilities are generated. In the centroblasts, the method of somatic hypermutation is activated, which introduces somatic mutations at a very high fee into rearranged Ig V genes. In some cases, intraclonal diversity of V area genes was noticed, indicating ongoing somatic hypermutation during clonal enlargement. However, several cases were identified that lacked Ig V gene rearrangements and that showed clonal T-cell receptor gene rearrangements. HodgkinLymphomaCellLines Tumor cell traces are priceless instruments for detailed genetic, biochemical, and practical studies of a malignancy. However, later studies confirmed that this line represents a cell tradition contamination. These cells are defined as uncommon cells that have a particular proliferative potential and that maintain the tumor clone, whereas the majority of the tumor clone lacks the potential to regrow to a full tumor.
Buy discount singulair 10 mg
Lineage-specific signaling in addition to transcription issue expression is managed by a network of cytokines and cytokine receptors asthma 6 steps singulair 5 mg buy generic on line. Depending on the kind of underlying neoplasm asthma treatment vs copd singulair 10 mg discount visa, eosinophil chemotactic cytokines may also be produced in an autocrine method. During an acute or chronic inflammatory course of, larger numbers of eosinophils may be recruited actively into local tissue websites. For example, mast cells provide the immediate stimuli upon activation to initiate allergic inflammation, leading to eosinophil recruitment through the later phases following an allergen encounter. In addition, mast cells and eosinophils are often elevated in numbers in allergic reactions, but in addition in nonallergic inflammatory situations and even in neoplastic states. Mast cells and eosinophils also work together with one another through cell�cell contact and through a quantity of different soluble mediators. Likewise, mast cell-derived heparin binds and stabilizes numerous chemokines, together with the eotaxins. In addition, mast cells are a supply of various cytokines involved in the regulation of eosinophil adhesion, migration, and function. Therefore, mast cells and eosinophils may usually appear as a dual target cell population, each in reactive and neoplastic states, which is a important level when contemplating the development of specific therapies. In case of atypical (immature) cells or questionable results, immunophenotyping could be performed to confirm the presence of eosinophils. In tissue sections, the quantification of eosinophils is a harder task that makes routine scientific evaluations considerably impractical. Alternative approaches corresponding to analysis of tissue secretions from affected organs. Indeed, these antigens might serve as reliable biomarkers of eosinophil involvement and activation in allergic, parasitic, and inflammatory diseases. In addition, therapy responses may be demonstrated and measured with these assays. The most incessantly detected molecular abnormality is the F/P fusion gene, a mutant gene created by an 800-kb interstitial deletion on chromosome 4q12. Although multiple organ techniques are concerned and the manifestation patterns are quite complex with varying programs and outcomes, some common pathogenetic components have been described. One frequent feature is the mobilization of the tissue microenvironment by eosinophil-derived mediators and cytokines, which finally ends up in tissue remodeling, fibrosis, and elevated angiogenesis. Another frequent factor is tissue inflammation, which is typically triggered by eosinophil-derived cytokines and chemokines. A variety of completely different compounds produced by eosinophils, including cationic proteins and numerous cytokines, might induce fibrosis. All these cytokines are produced and secreted by normal/reactive and in addition by neoplastic eosinophils. However, neoplastic eosinophils produce some of these cytokines in excess over their normal counterparts. An further clarification may be that eosinophil-derived mediators are toxic to sure (tissue-specific) cell sorts such as cardiomyocytes. Indeed, as famous earlier, eosinophils categorical numerous completely different cationic proteins able to inducing endothelial and endocardial harm and neurotoxicity. In addition, the eosinophil has the capability to generate reactive oxidative species that may increase tissue damage. These sufferers must be adopted closely, as they may progress to an overt eosinophil illness (neoplasm) over time. In uncertain instances, troponin levels and B-type natriuretic peptide ranges could also be helpful parameters. An endomyocardial biopsy could additionally be required in a few of these sufferers in order to set up the definitive diagnosis, but biopsies are normally not carried out in daily follow. The presence of eosinophils within the myocardium is at all times irregular and highly suggestive of eosinophil-mediated organopathy. Yes No Other molecular abnormality, clonal eosinophils, and/or elevated marrow blasts (5-19%) In addition, morphologic and histopathologic standards are used to arrive at a final prognosis relating to the underlying illness. These embody (1) an initial acute necrotic stage of brief duration (a few weeks) involving lively endomyocarditis, (2) a later thrombotic stage (several months) with mural thrombus formation over endocardial lesions, and (3) a late fibrotic stage (after approximately 2 years of illness) with growth of endomyocardial fibrosis. Echocardiography and angiography could fail to detect abnormalities at this early stage as a end result of ventricular thickening has not but occurred and endomyocardial biopsies, usually from the best ventricle, are required to make the right analysis of cardiac involvement. In the second stage, thrombi kind over the damaged endocardium in either of the ventricles or the atrium, typically with sparing of the aortic and pulmonary valves. Progressive scarring at websites of mural thrombus formation finally leads to the late fibrotic stage, with endomyocardial fibrosis leading to a restrictive cardiomyopathy and mitral or/and tricuspid valve regurgitation (see Table 71. The more frequent medical manifestations within the later progressive phases of endomyocardial fibrosis embody dyspnea, chest pain, indicators of left or right ventricular congestive coronary heart failure, or each, murmurs from mitral valve regurgitation, cardiomegaly, and T-wave inversions. The first form of neurologic involvement is brought on by thromboemboli, which can originate from intracardiac thrombi in the left ventricle but may develop regionally in cerebral vessels. Some of these thromboembolic episodes could occur before an overt cardiac manifestation has been documented. Patients with thrombotic complications might experience embolic strokes or transient ischemic assaults that might be a number of and recurrent, and these episodes may happen although the patient is satisfactorily anticoagulated. These sufferers could variably exhibit modifications in behavior, confusion, ataxia, and lack of memory. This contains symmetric or asymmetric sensory polyneuropathies, including sensory deficits, painful paresthesias, or combined sensory and motor defects (see Table 71. These neuropathies may enhance with corticosteroid administration or other remedies, and could also be secure or continue to progress regardless of remedy, or might improve and even resolve with time. The histopathology of the involved nerves normally shows varying levels of axonal loss, without evidence of vasculitis or direct or peripheral eosinophil infiltration. As within the coronary heart, several completely different eosinophil-derived (cytotoxic and stroma-targeting) mediators and cytokines may act collectively to induce tissue harm and tissue transforming, in addition to fibrosis and thrombosis. Some of the pulmonary features may also replicate modifications secondary to congestive coronary heart failure. Transudative pleural effusions are largely seen in patients with frank congestive heart failure. Pulmonary fibrosis is rare, however can develop in patients with endomyocardial fibrosis. In these sufferers, the cutaneous lesions fall into three categories: (1) angioedematous and urticarial lesions; (2) erythematous, pruritic papules and nodules; and (3) mucosal ulcerations (see Table seventy one. These patients have a dysfunction with recurrent attacks of angioedema and urticaria accompanied by fever and weight achieve. Oral sodium cromoglycate (cromolyn sodium), administered earlier than meals, has also been reported to be efficacious, although neither dapsone nor cromolyn is ready to reduce eosinophil counts within the blood. These lesions can appear at a number of websites, including the mouth, nose, pharynx, penis, esophagus, abdomen, and anus.
Buy singulair 4 mg visa
Therefore asthma treatment other than inhaler generic singulair 5 mg amex, the thalassemia patient born in the present period may count on to spend the majority of their life as an grownup asthma movie buy singulair 5 mg free shipping. There is at present restricted expertise among adult hematologists in the management of adult thalassemia patients. Transition plans are essential for all pediatric sufferers as they enter adulthood, so that they may receive age-appropriate care. Transition is very essential in thalassemia, as they require the regular uninterrupted schedule of 562 PartV RedBloodCells transfusions and chelation monitoring. Future efforts must be initiated to enhance the care of adult sufferers with thalassemia, including understanding the pathology of the disease and coaching new generations of hematologists with an understanding of thalassemia syndromes. Currently, the commonest cause of mortality in adults with thalassemia is related to cardiac disease and arrhythmias. These patients also have other continual diseases including cardiomyopathy, hepatic illness, endocrinopathies, osteoporosis, hypogonadism, pulmonary hypertension, and ongoing infectious danger from splenectomies. Regular observe up with an inside drugs primary care doctor can additionally be beneficial to make sure adults are conscious of current screening and treatment tips applicable to the overall population, for example age-appropriate most cancers screening, an space the place adult sufferers might not have beforehand been referred due to traditionally shorter lifespans. ExperimentalTherapies Enhancement of and Gene Expression Active -globin genes are hypomethylated in utero however are methylated and inactive after birth. Hypomethylation of the -globin genes may be induced by the drug 5-azacytidine; indeed, short-term administration of this drug produced the expected effect in vivo. Preparatory chemotherapeutic regimens to enhance immunosuppression and eradicate thalassemic clones utilizing hydroxyurea, azathioprine, fludarabine, busulfan, and cyclophosphamide have increased the survival fee of class 3 patients to 93%; the rejection fee fell to 8%. A phase I human gene therapy medical trial for hemoglobinopathies was initiated in France in 2007. However, the therapeutic Hb on this patient contributed only one-third of the whole Hb synthesized; the embryonic and fetal Hbs accounted equally for the remaining Hb. Multiple medical trials are currently underway using both gene therapy and genome enhancing technologies as healing approaches for thalassemia main. ThalassemiaIntermedia Approximately 10% of patients with homozygous -thalassemia exhibit a phenotype characterized by intermediate hematologic severity. For instance, homozygous -thalassemia in African Americans, Portuguese, and other populations could additionally be relatively mild, at least for the first twenty years of life. In other phrases, distinguishing between thalassemia major and thalassemia intermedia, which in turn is the excellence between initiating a regular transfusion program or not, requires consideration of the Hb stage and the standard of life (Table forty. Some nontransfused patients have regular growth and sexual growth, few medical issues, and regular or near-normal survival charges. However, others develop disfiguring facial modifications, markedly delayed progress and sexual maturation, heart failure, severe osteoporosis, repeated fractures, arthritis, and massive splenomegaly. Nonetheless, many families and physicians are reluctant to provoke a continual transfusion program because of concern about the dangers of long-term transfusion therapy and the inevitable want for iron chelation remedy. The need for normal transfusions typically develops in adults with thalassemia intermedia due to a further decline in the Hb level or a growing intolerance of the anemia. By the third or fourth decade, the entire iron burden could attain the degrees seen in transfusion-dependent sufferers. Deferoxamine, 564 PartV RedBloodCells deferiprone, and deferasirox have all proved to be protected and efficient in thalassemia intermedia. A hypercoagulable state may also contribute to the pulmonary hypertension that commonly happens in sufferers with thalassemia intermedia and is the first cause of congestive heart failure. This complication occurs less regularly in sufferers with thalassemia major because of the partial suppression of erythropoiesis by common transfusions. Masses of extramedullary hematopoietic tissue develop within the spinal epidural house, thorax, skull, pelvis, and elsewhere. For example, patients with extramedullary hematopoietic plenty could develop paraplegia from spinal twine compression or loss of visible acuity or visible fields attributable to optic nerve compression. The Hb level averages 1 or 2 g/dL decrease than that seen in regular individuals of the same age and gender. Hb F levels decline more slowly than traditional within the first yr of life, and the diagnostic elevated Hb A2 levels are established by roughly 6 months of age. Because iron deficiency may happen during pregnancy, iron supplementation has been advised to keep away from compounding the causes of anemia. Studies have instructed there could also be an increased tendency for gallstones and cholecystitis, however otherwise this situation should be largely asymptomatic. However, the gene deletions answerable for the commonest varieties are readily detectable by molecular biology methods. In general, partial deletions are extra deleterious and create a extra severe phenotype than full deletions. Nondeletional types of -thalassemia, which account for 15% to 20% of sufferers, arise from mutations just like those described for -thalassemia. The Quong Sze -globin chain (125LeuPro) is exceedingly labile and is destroyed so quickly after its synthesis that no Hb tetramers containing the mutant chain could be formed. Hemoglobin Constant Spring is an -globin chain variant synthesized in such small quantities (1�2% of normal) that it has the phenotypic influence of a severe nondeletion -thalassemia allele; nevertheless, the cs allele is all the time linked to a functioning -globin gene, so it has by no means been related to hydrops fetalis. During the newborn interval, small amounts (3%) of Hb Bart (4) can be seen by electrophoresis or other strategies. This condition is most frequently recognized when an apparently normal individual turns into the father or mother of a kid with Hb H disease after mating with a person with �-thalassemia trait. The mild extra of -globin chains might be removed in erythroblasts by proteolysis. At the molecular degree, +thalassemia has been found to be associated with two widespread gene deletions resulting from totally different nonhomologous crossing-over occasions between the 2 linked -globin genes: a three. Non-deletional HbH occurs when two deletional alpha(0) mutations occur with an alpha (+) mutation. Nondeletional Hb H genotypes usually tend to have larger proportion Hb H, extra splenomegaly, and more superior disease. Affected fetuses are usually born prematurely and are either stillborn or die shortly after start. These morphologic abnormalities and a unfavorable Coombs check outcome exclude hemolytic diseases caused by blood group incompatibility. A minor element recognized as Hb Portland (22) migrating in the place of Hb A can be seen. Extreme extramedullary erythropoiesis occurs in response to the profound hypoxia and hemolytic anemia characteristic of this disease. The universal edema characteristic of the hydrops fetalis syndrome is a reflection of severe congestive coronary heart failure and hypoalbuminemia in utero. This is partly a consequence of anemia, however the strikingly abnormal oxygen affinity of the tetrameric Hb Bart might be the most important determinant of the extreme tissue hypoxia. The oxygen dissociation curve of Hb Bart lacks the conventional sigmoid kind due to noncooperativity throughout oxygen loading and unloading and is markedly shifted to the left. The shift is so nice that little oxygen is released under conditions of low oxygen concentration in the tissues.
